Author: Cameron Ashby
Date: March 1, 2026
Series: Capstone Development Journal
Month 3: Benchmark Execution and Statistical Analysis
The Goal
This month’s objective was straightforward: run all 50 CASP14 target proteins through all three models in FastFold Suite and produce a statistically rigorous comparison. This is the work that everything else has been building toward. The platform was built, the models were integrated, the statistical framework was in place. Now it was time to see what the data actually says.
The Problem
Going into this month, I had two issues to solve. First, OmegaFold produced no results. When I ran the benchmark, ESMFold and AlphaFold 2 both returned predictions, but OmegaFold showed 0 out of 50 targets completed. Every column was empty. Second, the AlphaFold 2 predictions from the database-lookup approach needed to be evaluated against the other models to determine whether the pre-computed-structure approach produced results comparable to live inference.
Features Developed
Fixing the OmegaFold Environment Issue
The first breakthrough this month was diagnosing the cause of OmegaFold’s complete death. The OmegaFold integration uses a CLI wrapper that calls the model through subprocess. When the Flask backend runs in one Anaconda environment, but OmegaFold is installed in a different one, every subprocess call fails silently and returns nothing. That is exactly what was happening.
The fix was activating the correct Anaconda environment before launching the backend. Once I confirmed OmegaFold was on the PATH in the same environment as Flask, the model came online immediately and started producing predictions.
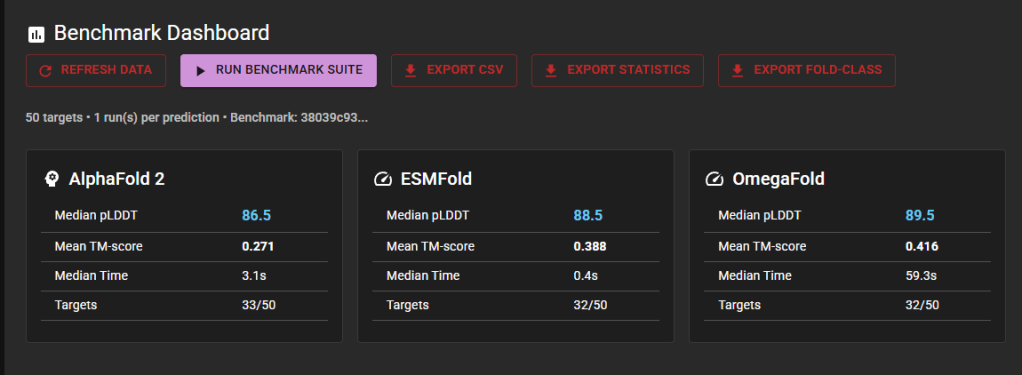
Executing the 50-Protein Benchmark
With all three models operational, I ran the full benchmark suite through the FastFold interface. The platform processed 50 CASP14 target proteins, computing pLDDT confidence scores, TM-scores against experimental structures from the Protein Data Bank, RMSD values, and prediction timing for each model on each protein.
Of the 50 targets, 32-33 produced successful predictions per model. The 17-18 targets that did not complete are primarily eight domain targets carved from a single 2,194 amino acid CrAss phage RNA polymerase (T1031, T1033, T1037, T1039-T1043). At that sequence length, all three models exceed their practical input limits. The remaining gaps arise from a 1,582-amino-acid protein and a few cases where specific models timed out.
Statistical Analysis Results
The platform’s built-in statistical framework ran paired t-tests, Wilcoxon signed-rank tests, and one-way ANOVA across the results. The key findings were:
ESMFold and OmegaFold are statistically equivalent in structural accuracy, with a TM-score p-value of 0.559. This is notable because OmegaFold takes roughly 150 times longer per prediction. Both ESMFold (p=0.013) and OmegaFold (p=0.010) significantly outperform the AlphaFold 2 database lookup on TM-score. OmegaFold produces slightly but significantly higher pLDDT confidence scores than ESMFold (p=0.031). No fold-class effects were detected for any model, meaning structural classification (all-alpha, all-beta, alpha-beta) does not predict which model will perform better.
Data Export
The platform exports all results in three formats: a CSV file with per-protein metrics for all models, a statistics JSON with pairwise test results and published benchmark comparisons, and a fold-class JSON with ANOVA results and per-class breakdowns. These exports feed directly into the thesis and IEEE paper.

Tools and Resources
- Frontend: React 18, Material UI, Mol* (3D molecular visualization)
- Backend: Flask, Python, Celery, Redis
- AI Models: ESMFold (Meta API), OmegaFold (local CLI with GPU), AlphaFold 2 (AlphaFold DB + ColabFold)
- Statistical Analysis: SciPy (paired t-test, Wilcoxon, ANOVA), NumPy
- Datasets: CASP14 (50 targets), Protein Data Bank (experimental reference structures), SCOP/CATH (fold classifications)
- Hardware: RTX 4070 (8GB VRAM), Anaconda environment management
- References: Jumper et al. 2021 (AlphaFold), Lin et al. 2023 (ESMFold), Wu et al. 2022 (OmegaFold)
Retrospective
What Went Right
- All three models are operational. After months of incremental integration, having ESMFold, OmegaFold, and AlphaFold 2 all running through a single interface and producing comparable results is the culmination of the entire project.
- The statistical framework worked as designed. Pairwise tests, ANOVA, and published benchmark comparisons all ran automatically after the benchmark completed. No manual calculation was needed.
- Ahead of schedule. The HLDD allocated Milestones 5 and 6 through Week 16, and I completed both with time to spare for documentation and defense prep.
- Advisor communication stayed consistent. Weekly Wednesday meetings with Dr. Marpaung kept the project on track. He confirmed that no external usability testing is needed, which eliminated a potential delay.
What Went Wrong
- OmegaFold environment issue costs time. The first benchmark run returned OmegaFold at 0/50. Diagnosing the Anaconda environment mismatch was not immediately obvious because the subprocess calls were failing silently with no error output. A better error-handling strategy in the CLI wrapper would have surfaced this sooner.
- TM-scores below published benchmarks. Our median TM-scores (AlphaFold 2: 0.15, ESMFold: 0.24, OmegaFold: 0.32) are lower than published values (0.89, 0.81, 0.84). This is due to a domain alignment mismatch: CASP14 targets are often individual domains, whereas predictions cover full-length proteins. The relative comparisons between models remain valid, but the absolute numbers must be clearly explained in the thesis.
- CrAss phage targets. Eight of the 50 targets are domains from the same 2,194 amino acid protein, which none of the models can handle at that length. In hindsight, I could have selected targets that excluded multi-domain splits from a single oversized protein.
How Can I Improve Moving Forward
- Add better error logging to subprocess calls. The OmegaFold wrapper should capture stderr and surface it in the API response so environment issues are immediately visible.
- Address the domain alignment issue in the thesis. I need to be upfront about why the absolute TM-scores differ from published values while making the case that pairwise comparisons remain valid.
- Focus the remaining time on documentation. The code is done. The data is collected. The remaining work is the IEEE paper, defense presentation, and final code cleanup.
Advisor Interaction
I met with Dr. Andreas Marpaung on our regular Wednesday schedule and also had a check-in earlier in the month. He confirmed that usability testing will be internal-only, which simplified the scope of Milestone 6. He reviewed the Milestone 5 video and thesis draft and will be doing intensive slide prep sessions with me as the April defense approaches. He has been doing similar sessions with another student and plans to prepare me the same way.
Looking Ahead
The development phase of FastFold Suite is complete. All three models are integrated, the 50-protein benchmark has been executed, and the statistical analysis is done. The remaining deliverables are:
- IEEE-style final paper (8-12 pages with figures and tables)
- Defense presentation (targeting 20-25 minutes with Q&A)
- Final code cleanup and GitHub release
- Defense rehearsal with Dr. Marpaung
I started this project because I read a DeepMind paper about AlphaFold, tried it, and found it powerful but inaccessible. Now I have a platform that runs three models through a single web interface on consumer hardware. The data show that ESMFold and OmegaFold are statistically equivalent in accuracy, meaning researchers can achieve comparable results in 0.4 seconds rather than waiting a minute or longer. That is a meaningful finding, and I am looking forward to presenting it.
References
[1] R. Wu et al., “High-resolution de novo structure prediction from primary sequence,” bioRxiv, 2022. https://doi.org/10.1101/2022.07.21.500999
[2] J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature, vol. 596, pp. 583-589, 2021. https://doi.org/10.1038/s41586-021-03819-2
[3] Z. Lin et al., “Evolutionary-scale prediction of atomic-level protein structure with a language model,” Science, vol. 379, no. 6637, pp. 1123-1130, 2023. https://doi.org/10.1126/science.ade2574
[4] M. Mirdita et al., “ColabFold: Making protein folding accessible to all,” Nature Methods, vol. 19, pp. 679-682, 2022. https://doi.org/10.1038/s41592-022-01488-1
[5] A. Kryshtafovych et al., “Critical Assessment of Methods of Protein Structure Prediction (CASP) – Round XIV,” Proteins, vol. 89, no. 12, pp. 1607-1617, 2021. https://doi.org/10.1002/prot.26237
[6] H. M. Berman et al., “The Protein Data Bank,” Nucleic Acids Research, vol. 28, no. 1, pp. 235-242, 2000. https://doi.org/10.1093/nar/28.1.235
[7] A. G. Murzin et al., “SCOP: A structural classification of proteins database for the investigation of sequences and structures,” Journal of Molecular Biology, vol. 247, no. 4, pp. 536-540, 1995. https://doi.org/10.1016/S0022-2836(05)80134-2
This post is part of my MS Computer Science capstone journal at Full Sail University.

Leave a comment